- 移动端


上海中科新生命生物科技有限公司
18 年
手机商铺
商家活跃:
产品热度:
- NaN
- 0
- 1
- 0
- 3
文献支持
16S/ITS/18S扩增子测序
询价




公司新闻/正文
项目文章STTT(IF 52.7)| 河北医科大学杨展/岳南希等揭示LPE18:1通过重编程脂质代谢影响肾癌进展机制
449 人阅读发布时间:2025-12-15 14:12
 透明细胞肾细胞癌(ccRCC)以细胞内异常脂质累积为典型特征,但其脂质累积的来源及驱动机制尚未明确。肾周脂肪(PAT)在肿瘤旁持续“褐变”,被推测是关键的外源性脂质来源,然而还缺乏直接分子证据。同时,溶血磷脂酰乙,醇胺(LPE)在ccRCC进展中的具体作用、其是否源自 PAT,以及如何调控并重塑ccRCC的脂质代谢,目前尚未明确。深入解析上述问题,有望系统揭示 ccRCC 的代谢调控规律,为该疾病的治疗提供全新潜在靶点。
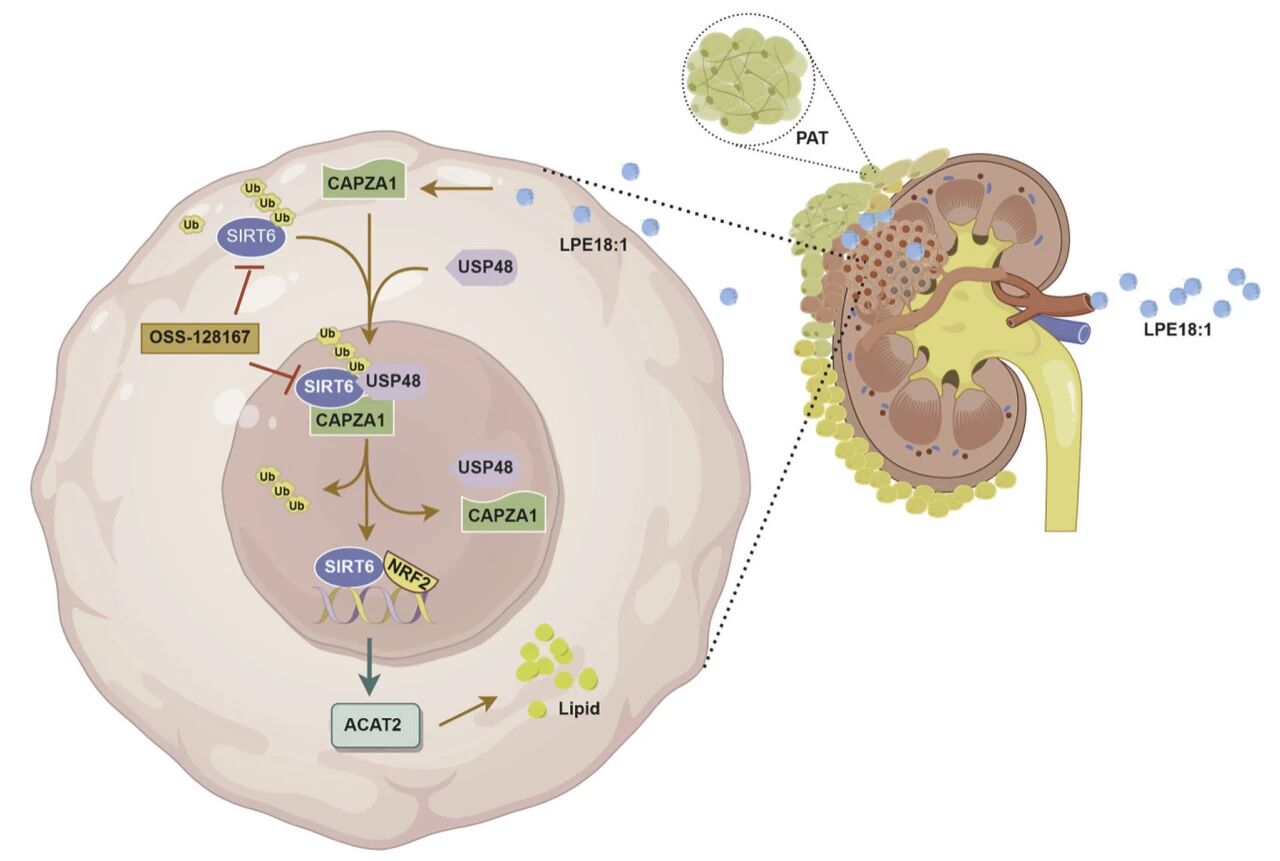
2025年12月8日,河北医科大学杨展/岳南希、中国医科大学陈小楠及石家庄市人民医院瞿长宝等团队在顶刊Signal Transduction and Targeted Therapy(IF52.7)在线发表了题为“Lysophosphatidylethanolamine 18:1 drives clear cell renal cell carcinoma by stabilizing SIRT6 to reprogram lipid metabolism”的研究性论文,揭示了PAT褐变分泌代谢物LPE18:1→激活CAPZA1→招募USP48稳定SIRT6→SIRT6/NRF2共激活ACAT2→胆固醇酯化→脂质沉积→透明细胞肾癌爆发,阻断CAPZA1/SIRT6轴即可“饿死”肿瘤。中科新生命为该研究提供了代谢组检测服务。
透明细胞肾细胞癌(ccRCC)以细胞内异常脂质累积为典型特征,但其脂质累积的来源及驱动机制尚未明确。肾周脂肪(PAT)在肿瘤旁持续“褐变”,被推测是关键的外源性脂质来源,然而还缺乏直接分子证据。同时,溶血磷脂酰乙,醇胺(LPE)在ccRCC进展中的具体作用、其是否源自 PAT,以及如何调控并重塑ccRCC的脂质代谢,目前尚未明确。深入解析上述问题,有望系统揭示 ccRCC 的代谢调控规律,为该疾病的治疗提供全新潜在靶点。
2025年12月8日,河北医科大学杨展/岳南希、中国医科大学陈小楠及石家庄市人民医院瞿长宝等团队在顶刊Signal Transduction and Targeted Therapy(IF52.7)在线发表了题为“Lysophosphatidylethanolamine 18:1 drives clear cell renal cell carcinoma by stabilizing SIRT6 to reprogram lipid metabolism”的研究性论文,揭示了PAT褐变分泌代谢物LPE18:1→激活CAPZA1→招募USP48稳定SIRT6→SIRT6/NRF2共激活ACAT2→胆固醇酯化→脂质沉积→透明细胞肾癌爆发,阻断CAPZA1/SIRT6轴即可“饿死”肿瘤。中科新生命为该研究提供了代谢组检测服务。

研究材料
ccRCC 组织、配对脂肪与血液、多株细胞系、裸鼠移植瘤
研究步骤
步骤1:发现肿瘤微环境代谢特征——发现PAT褐变,鉴定其特异性分泌LPE18:1,并被ccRCC细胞高效摄取,提示其为外源性肿瘤代谢燃料;
步骤2:阐明LPE18:1的致癌功能——LPE18:1促进ccRCC细胞增殖并驱动脂代谢重编程,形成“脂质沉积-能量供给”闭环;
步骤3:解析LPE18:1下游信号轴——LPE18:1上调CAPZA1表达,其高表达与患者不良预后相关。敲低CAPZA1可阻断LPE18:1诱导的增殖与脂质沉积,体内外验证其必要性。
步骤4:揭示CAPZA1下游机制——CAPZA1直接结合并稳定SIRT6:通过抑制泛素-蛋白酶体途径,并招募USP48对SIRT6去泛素化。
步骤5:阐明完整信号轴与转化价值。
PAT褐变 → LPE18:1分泌 → ccRCC摄取 → CAPZA1↑ → USP48介导SIRT6去泛素化 → SIRT6稳定 → 与NRF2激活ACAT2 → 脂滴堆积 → 肿瘤生长
研究结果
1. PAT组织褐变驱动LPE18:1产生,进而被ccRCC肿瘤利用
组织学与分子检测证实PAT呈多房状且高表达UCP1,表明其已褐变。代谢组学筛选发现,LPE18:1在PAT、肿瘤组织及肾静脉血中均特异性上调,是共同差异脂质。定量分析显示PAT中LPE18:1含量约为皮下脂肪的2.5倍,且肾动脉血浓度高于静脉血,提示LPE18:1是由PAT释放;但是肿瘤组织内LPE18:1水平又最低。体外实验证实ccRCC细胞对LPE18:1具有高效摄取能力(24h消耗>70%)。综上,PAT褐变产生并释放LPE18:1,被ccRCC肿瘤细胞主动摄取消耗。
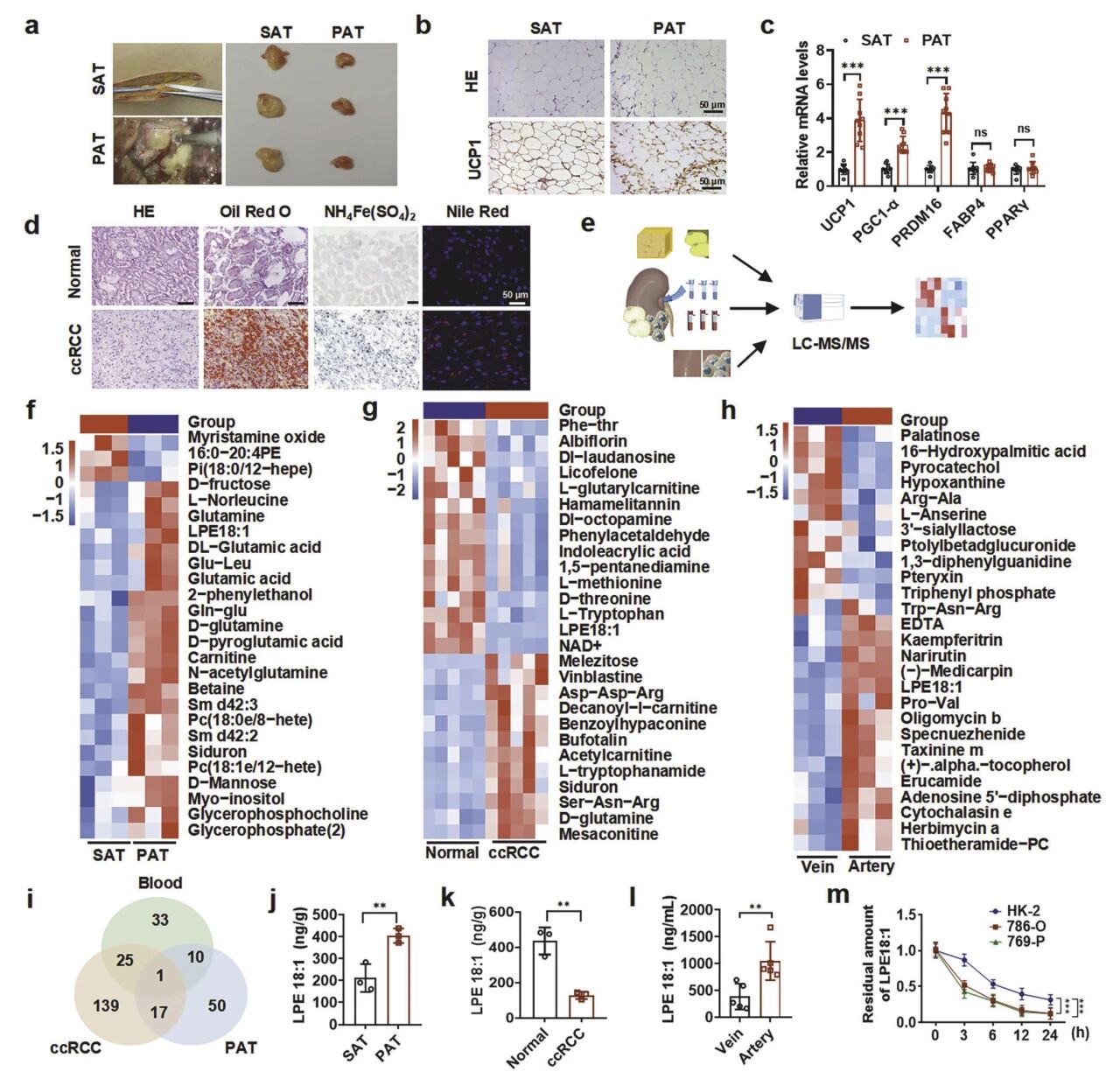 图2 肾周脂肪组织的褐变增强了LPE18:1的生成及ccRCC肿瘤对其的消耗
2. LPE18:1通过增强脂质沉积和线粒体能量产生促进ccRCC细胞增殖
LPE18:1(40 μM)可促进ccRCC细胞(786-O、769-P)增殖,显著提升EdU阳性率、集落形成能力及长期生长曲线,而对正常肾小管上皮细胞HK-2无影响。ccRCC细胞内出现大量脂滴,脂质定量证实总胆固醇和甘油三酯含量分别提升约1.8倍与2.2倍。Seahorse能量代谢分析进一步揭示,ccRCC细胞线粒体基础耗氧率与最大呼吸能力均显著增强,ATP产量提升约2倍,表明形成了“脂质沉积-能量供给”的代谢闭环。
图2 肾周脂肪组织的褐变增强了LPE18:1的生成及ccRCC肿瘤对其的消耗
2. LPE18:1通过增强脂质沉积和线粒体能量产生促进ccRCC细胞增殖
LPE18:1(40 μM)可促进ccRCC细胞(786-O、769-P)增殖,显著提升EdU阳性率、集落形成能力及长期生长曲线,而对正常肾小管上皮细胞HK-2无影响。ccRCC细胞内出现大量脂滴,脂质定量证实总胆固醇和甘油三酯含量分别提升约1.8倍与2.2倍。Seahorse能量代谢分析进一步揭示,ccRCC细胞线粒体基础耗氧率与最大呼吸能力均显著增强,ATP产量提升约2倍,表明形成了“脂质沉积-能量供给”的代谢闭环。
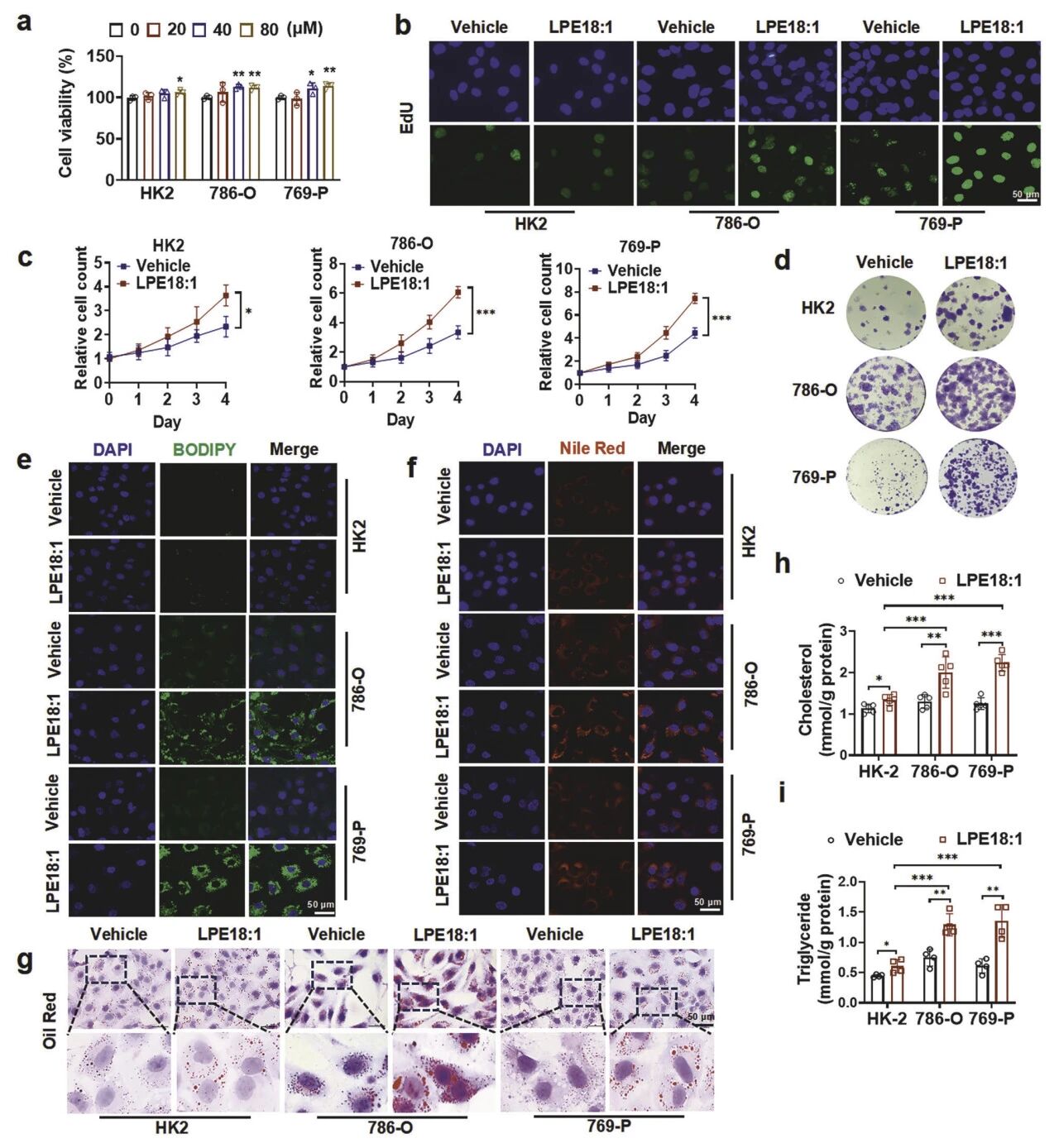 图3 LPE18:1促进ccRCC增殖与脂质积累
3. LPE18:1促进肾透明细胞癌中CAPZA1的表达
转录组测序发现LPE18:1能特异性上调ccRCC细胞中CAPZA1的表达(mRNA约4.3倍),该结果在转录与蛋白水平均获验证。临床样本显示肿瘤组织CAPZA1蛋白水平约为癌旁的2.6倍。TCGA数据库分析进一步证实,CAPZA1在肿瘤中表达显著升高(约2.2倍),其高表达与晚期TNM分期正相关,并提示更差的总体生存率(5年生存率降低38%,AUC=0.82)。这些结果表明,LPE18:1诱导的CAPZA1高表达是ccRCC的关键促癌因子,具备作为预后生物标志物的潜力。
图3 LPE18:1促进ccRCC增殖与脂质积累
3. LPE18:1促进肾透明细胞癌中CAPZA1的表达
转录组测序发现LPE18:1能特异性上调ccRCC细胞中CAPZA1的表达(mRNA约4.3倍),该结果在转录与蛋白水平均获验证。临床样本显示肿瘤组织CAPZA1蛋白水平约为癌旁的2.6倍。TCGA数据库分析进一步证实,CAPZA1在肿瘤中表达显著升高(约2.2倍),其高表达与晚期TNM分期正相关,并提示更差的总体生存率(5年生存率降低38%,AUC=0.82)。这些结果表明,LPE18:1诱导的CAPZA1高表达是ccRCC的关键促癌因子,具备作为预后生物标志物的潜力。
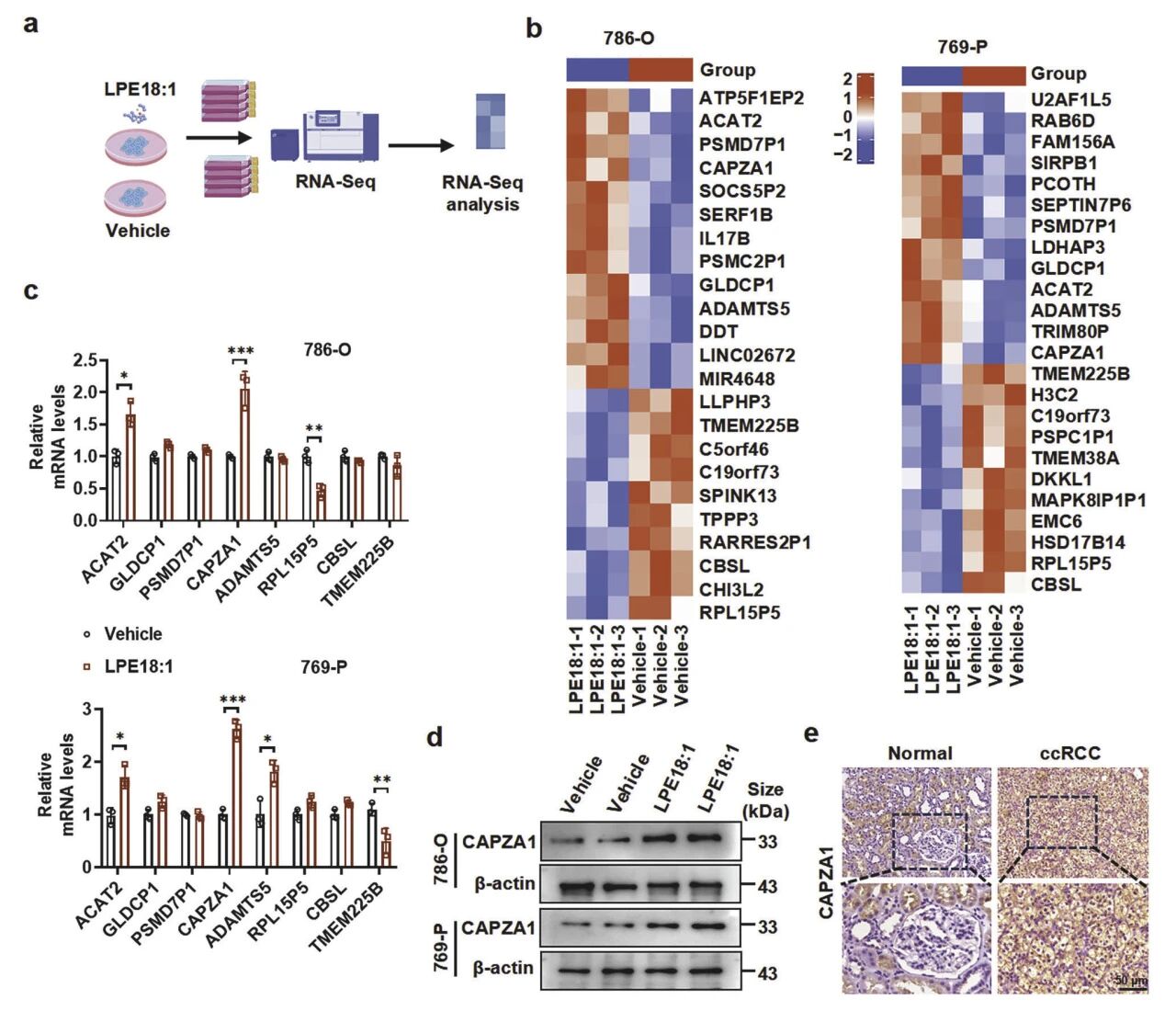 图4 LPE18:1 在肾透明细胞癌细胞和组织中诱导 CAPZA1 上调
4. CAPZA1是LPE18:1促进ccRCC增殖及脂质沉积的关键介质
LPE18:1能显著促进肿瘤体积增大、加速生长曲线,并提高组织内细胞密度;而敲除CAPZA1则可明显抑制上述促瘤效应,联合处理组肿瘤体积显著小于LPE18:1单独处理组。免疫组化与免疫荧光分析进一步表明,LPE18:1上调细胞周期蛋白CDK4及增殖标志物Ki-67的表达,而CAPZA1敲除则降低二者表达,证实其抑制细胞周期进展与增殖的作用。脂质沉积染色显示,LPE18:1诱导肿瘤内脂滴大量积累,该现象可被CAPZA1敲除几乎完全阻断。
图4 LPE18:1 在肾透明细胞癌细胞和组织中诱导 CAPZA1 上调
4. CAPZA1是LPE18:1促进ccRCC增殖及脂质沉积的关键介质
LPE18:1能显著促进肿瘤体积增大、加速生长曲线,并提高组织内细胞密度;而敲除CAPZA1则可明显抑制上述促瘤效应,联合处理组肿瘤体积显著小于LPE18:1单独处理组。免疫组化与免疫荧光分析进一步表明,LPE18:1上调细胞周期蛋白CDK4及增殖标志物Ki-67的表达,而CAPZA1敲除则降低二者表达,证实其抑制细胞周期进展与增殖的作用。脂质沉积染色显示,LPE18:1诱导肿瘤内脂滴大量积累,该现象可被CAPZA1敲除几乎完全阻断。
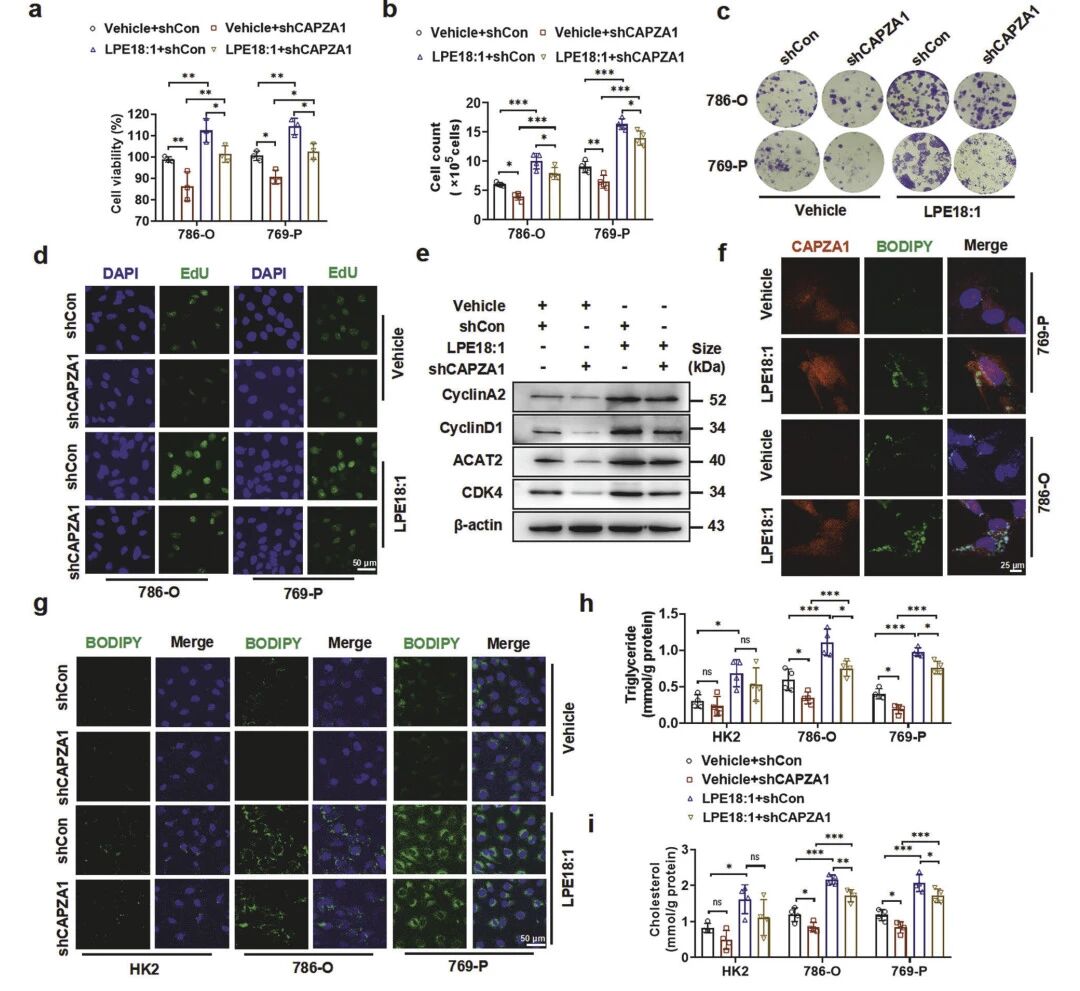 图5 LPE18:1诱导的ccRCC增殖与脂质沉积依赖于CAPZA1
5. 敲低CAPZA1可抑制LPE18:1驱动的ccRCC体内成瘤及脂质积累
体内实验证实,敲低CAPZA1完全阻断了LPE18:1对ccRCC肿瘤的促生长作用。在裸鼠皮下成瘤模型中,LPE18:1使对照瘤体积增大约2.5倍、重量达0.82 g,而敲低CAPZA1后瘤体积与重量均恢复至基线。同时逆转了LPE18:1诱导的高细胞密度和CDK4、Ki-67高表达等表型,并使其促脂质沉积效应基本消失(脂滴面积从28%降至6%),胆固醇/甘油三酯下降。结果表明,CAPZA1是LPE18:1驱动体内肿瘤生长与脂质沉积的必需介质。
图5 LPE18:1诱导的ccRCC增殖与脂质沉积依赖于CAPZA1
5. 敲低CAPZA1可抑制LPE18:1驱动的ccRCC体内成瘤及脂质积累
体内实验证实,敲低CAPZA1完全阻断了LPE18:1对ccRCC肿瘤的促生长作用。在裸鼠皮下成瘤模型中,LPE18:1使对照瘤体积增大约2.5倍、重量达0.82 g,而敲低CAPZA1后瘤体积与重量均恢复至基线。同时逆转了LPE18:1诱导的高细胞密度和CDK4、Ki-67高表达等表型,并使其促脂质沉积效应基本消失(脂滴面积从28%降至6%),胆固醇/甘油三酯下降。结果表明,CAPZA1是LPE18:1驱动体内肿瘤生长与脂质沉积的必需介质。
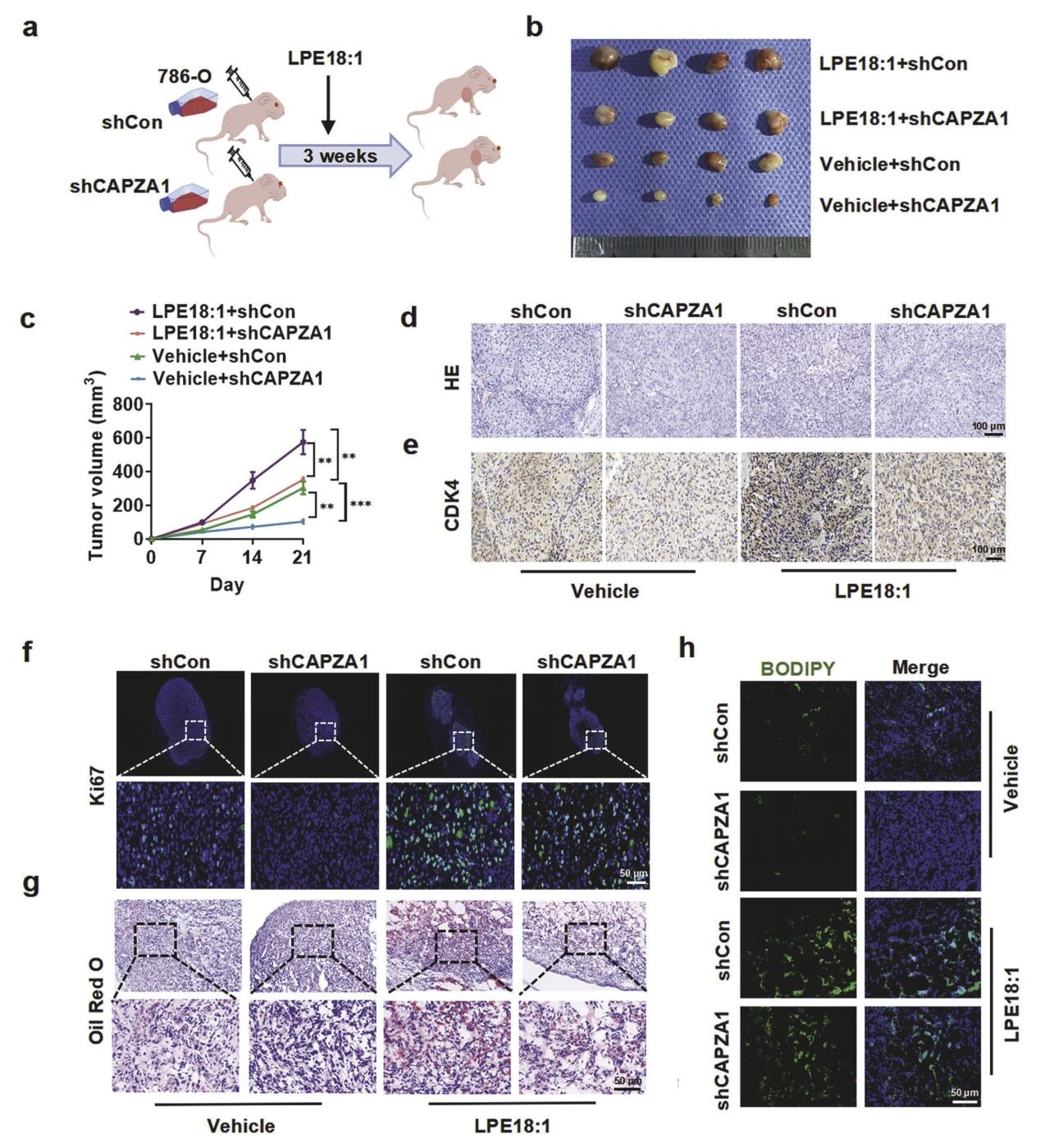 图6 CAPZA1缺失在体内减弱了LPE18:1诱导的肿瘤生长与脂质沉积
6. 在ccRCC中,SIRT6是CAPZA1的关键下游效应分子
互作蛋白筛选证实SIRT6是CAPZA1的关键下游效应分子。敲低CAPZA1会削弱与SIRT6的互作;计算模拟与Co-IP显示二者可直接结合,且LPE18:1增强该互作。免疫荧光证实二者在胞浆-核周共定位,且依赖于CAPZA1。临床样本中,SIRT6蛋白在肿瘤中高表达且与CAPZA1强正相关。体内实验也表明,敲低CAPZA1显著降低肿瘤内SIRT6蛋白水平。
图6 CAPZA1缺失在体内减弱了LPE18:1诱导的肿瘤生长与脂质沉积
6. 在ccRCC中,SIRT6是CAPZA1的关键下游效应分子
互作蛋白筛选证实SIRT6是CAPZA1的关键下游效应分子。敲低CAPZA1会削弱与SIRT6的互作;计算模拟与Co-IP显示二者可直接结合,且LPE18:1增强该互作。免疫荧光证实二者在胞浆-核周共定位,且依赖于CAPZA1。临床样本中,SIRT6蛋白在肿瘤中高表达且与CAPZA1强正相关。体内实验也表明,敲低CAPZA1显著降低肿瘤内SIRT6蛋白水平。
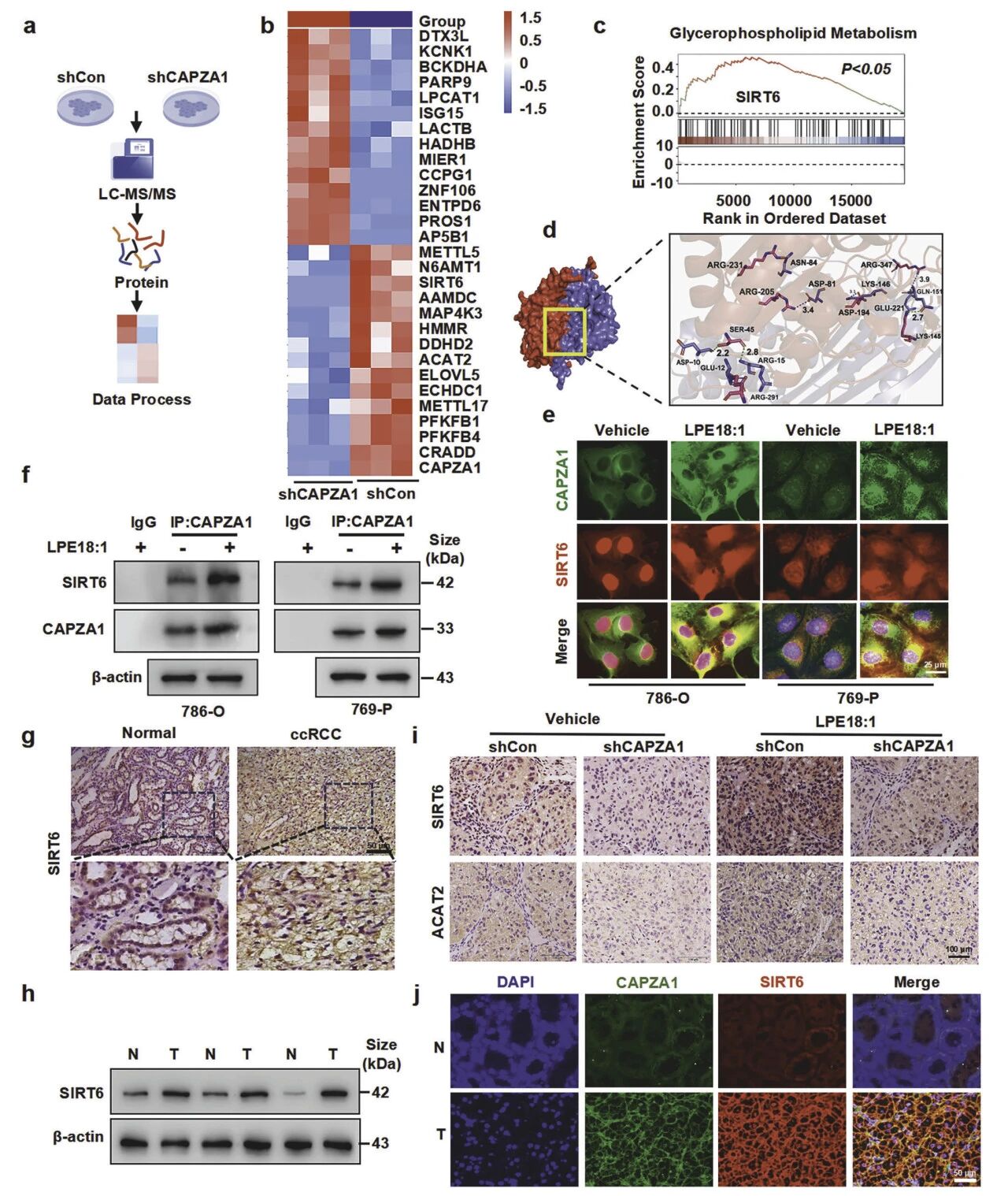 图7 SIRT6是ccRCC中CAPZA1的关键下游效应分子
7. LPE18:1诱导的细胞生长及脂质积累在体外和体内均依赖于SIRT6
SIRT6是LPE18:1/CAPZA1信号轴下游调控ccRCC增殖与脂质沉积的关键执行分子。敲低SIRT6可完全阻断LPE18:1诱导的增殖与脂滴积累,表型与敲低CAPZA1一样;二者均能同等抑制ACAT2与CDK4上调,且双敲无叠加,证实位于同一线性通路。SIRT6抑制剂OSS-128167可抑制ACAT2表达与脂质沉积,并逆转CAPZA1过表达的表型。体内实验表明,抑制SIRT6能使原位瘤体积减小52%,且与CAPZA1敲低具有协同抑瘤作用。
图7 SIRT6是ccRCC中CAPZA1的关键下游效应分子
7. LPE18:1诱导的细胞生长及脂质积累在体外和体内均依赖于SIRT6
SIRT6是LPE18:1/CAPZA1信号轴下游调控ccRCC增殖与脂质沉积的关键执行分子。敲低SIRT6可完全阻断LPE18:1诱导的增殖与脂滴积累,表型与敲低CAPZA1一样;二者均能同等抑制ACAT2与CDK4上调,且双敲无叠加,证实位于同一线性通路。SIRT6抑制剂OSS-128167可抑制ACAT2表达与脂质沉积,并逆转CAPZA1过表达的表型。体内实验表明,抑制SIRT6能使原位瘤体积减小52%,且与CAPZA1敲低具有协同抑瘤作用。
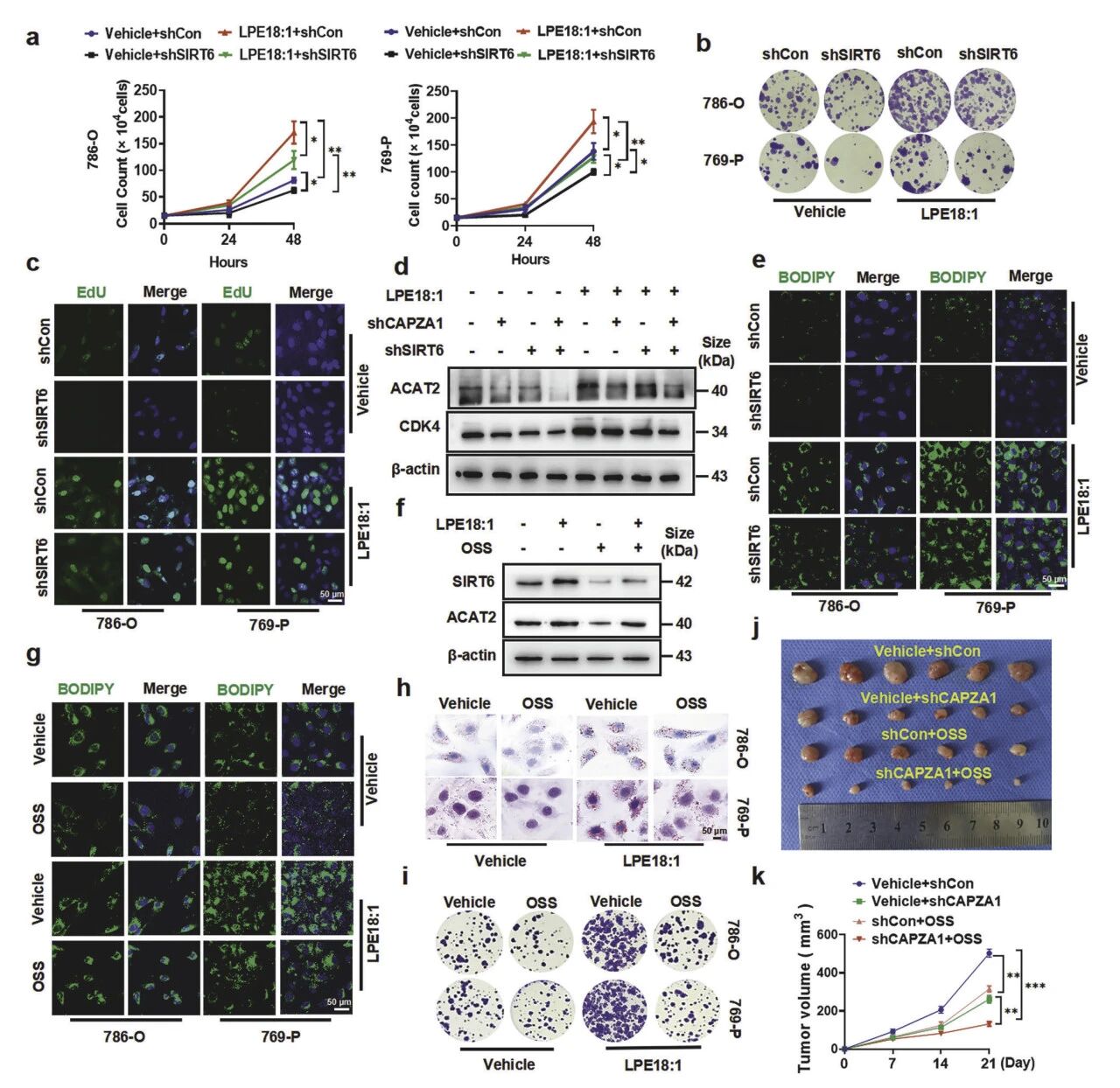 图8 SIRT6是LPE18:1/CAPZA1驱动ccRCC增殖与脂质沉积的关键因子
8. CAPZA1通过抑制泛素蛋白酶体途径增强SIRT6的稳定性
敲低或过表达CAPZA1可导致SIRT6蛋白水平下降70%或上升2.5倍,而mRNA无变化,表明其为翻译后调控。放线,菌酮(CHX)追踪显示,敲低CAPZA1使SIRT6半衰期从12h缩短至4h,过表达则延长至24h以上,证明CAPZA1抑制其降解。进一步研究,蛋白酶体抑制剂MG132可完全逆转CAPZA1敲低引起的SIRT6下降,且敲低CAPZA1使SIRT6多聚泛素化增加3.2倍,明确其通过抑制泛素-蛋白酶体途径稳定SIRT6。
图8 SIRT6是LPE18:1/CAPZA1驱动ccRCC增殖与脂质沉积的关键因子
8. CAPZA1通过抑制泛素蛋白酶体途径增强SIRT6的稳定性
敲低或过表达CAPZA1可导致SIRT6蛋白水平下降70%或上升2.5倍,而mRNA无变化,表明其为翻译后调控。放线,菌酮(CHX)追踪显示,敲低CAPZA1使SIRT6半衰期从12h缩短至4h,过表达则延长至24h以上,证明CAPZA1抑制其降解。进一步研究,蛋白酶体抑制剂MG132可完全逆转CAPZA1敲低引起的SIRT6下降,且敲低CAPZA1使SIRT6多聚泛素化增加3.2倍,明确其通过抑制泛素-蛋白酶体途径稳定SIRT6。
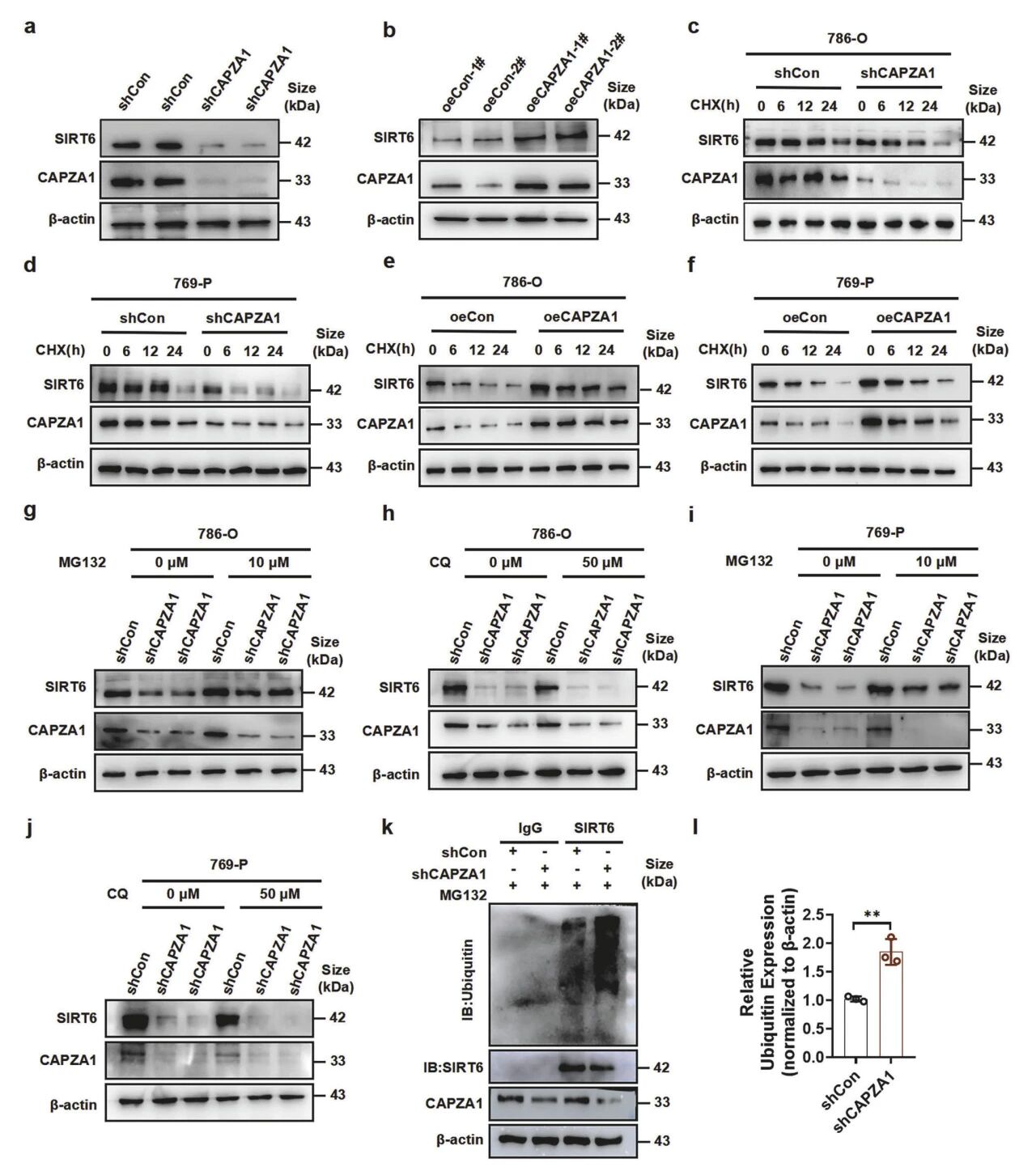 图9 CAPZA1通过泛素-蛋白酶体途径增强SIRT6的稳定性
9. CAPZA1通过介导USP48与SIRT6的相互作用来促进SIRT6的稳定
CAPZA1作为必需的“桥梁”,其存在是USP48与SIRT6结合并实现共定位的前提;敲低CAPZA1会显著削弱USP48与SIRT6的互作。敲低USP48会导致SIRT6蛋白下降、半衰期缩短,且该效应可被MG132逆转,证明USP48通过抑制泛素-蛋白酶体降解稳定SIRT6。进一步研究表明,LPE18:1或过表达CAPZA1能促进SIRT6-USP48结合并减少SIRT6泛素化;反之,敲低CAPZA1或USP48则增强SIRT6泛素化。
图9 CAPZA1通过泛素-蛋白酶体途径增强SIRT6的稳定性
9. CAPZA1通过介导USP48与SIRT6的相互作用来促进SIRT6的稳定
CAPZA1作为必需的“桥梁”,其存在是USP48与SIRT6结合并实现共定位的前提;敲低CAPZA1会显著削弱USP48与SIRT6的互作。敲低USP48会导致SIRT6蛋白下降、半衰期缩短,且该效应可被MG132逆转,证明USP48通过抑制泛素-蛋白酶体降解稳定SIRT6。进一步研究表明,LPE18:1或过表达CAPZA1能促进SIRT6-USP48结合并减少SIRT6泛素化;反之,敲低CAPZA1或USP48则增强SIRT6泛素化。
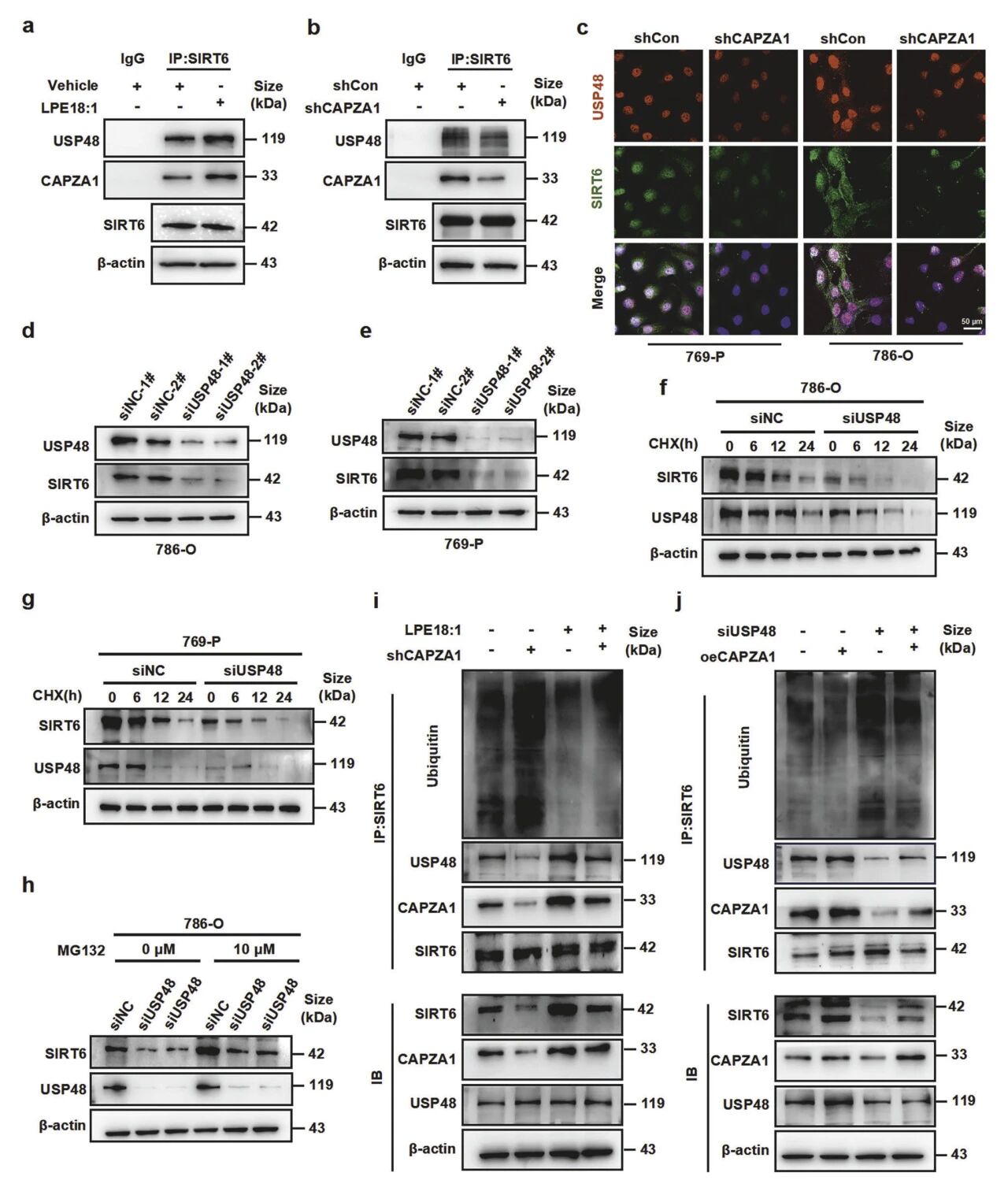 图10 CAPZA1招募USP48以对SIRT6进行去泛素化
图10 CAPZA1招募USP48以对SIRT6进行去泛素化
总结
该研究揭示了一种新的信号轴,将SIRT6介导的表观遗传调控与脂质代谢重编程联系起来,为PAT如何驱动ccRCC侵袭性提供了机制见解,并确定了CAPZA1/SIRT6作为ccRCC和其他相关疾病管理的潜在治疗靶点。
中科优品推荐 为满足前沿研究对定量脂质数据的高标准要求,中科新生命对标 CNS 等顶刊所采用的脂质分析策略,在成熟的HL2400高通量靶向脂质组基础上,全面升级成HL6000高通量靶向脂质组。该产品基于AB SCIEX 7500+高性能质谱仪,具备高通量、高灵敏度的技术优势,可一次性实现近60种脂质亚类、约6000种脂质分子的检测。同时采用双色谱柱分离技术,结合数十种同位素内标进行辅助定性与定量,能够全面、可靠地揭示生物体内的脂质代谢状态。针对不同样本类型,我们提供专属检测方案。欢迎各位老师垂询合作!

关于中科新生命
上海中科新生命生物科技有限公司(APTBIO)创立于 2004 年,由原中国科学院上海生命科学研究院蛋白质组研究中心孵化而来,是国内质谱多组学应用领域的开拓者。公司以 “AI + 质谱多组学” 双核驱动创新,构建智能化组学生态。拥有自主知识产权的质谱检测平台与 AI 大数据分析系统,聚焦科技服务、生物医药及大健康消费三大领域,为全球科研机构、医院、药企提供从基础研究到临床转化的一站式解决方案。融合多组学技术与人工智能,围绕生物标志物发掘、药物靶点筛选及个性化诊疗等方向,构建具有国际竞争力的组学数据库与算法模型,推动转化医学进程,加速创新药物研发,成为推动生命科学数字化升级的核心引领者。